Chapter 2
WHAT IS DEMENTIA
The World Health Organisation describes dementia as syndromes where cognitive function (the ability to process thought) deteriorates beyond what might be seen with normal ageing. Dementia syndromes are chronic and progressive. In the early stages, people may not realise dementia is developing, or they dismiss their symptoms as a normal part of ageing. In some forms of dementia, people do not have insight into their own deficits at all.
A common misconception is that dementia is one disease. In fact, dementia is an umbrella term for several different diseases that each lead to the loss of mental abilities because of the progressive loss of neurons (brain cells) and their function.
Another misconception is that dementia just relates to memory loss. In fact, the diseases that cause dementia can affect different people in different ways. Dementia can impact a person’s memory, thinking, orientation, comprehension, judgement, language, personality, emotional control and behaviour. These symptoms affect a person’s life and, as the disease progresses, their ability to live independently.


The lesion shows up as a deficit on a functional scan looking at glucose metabolism in the brain (FDG-PET scan). On this PET scan, thick arrows point to reduced areas of glucose metabolism at the back of the brain with relatively preserved metabolism at the front of the brain (thin arrows).
Common dementia types
Dementia symptoms usually evolve gradually. The earliest signs are often recognised in hindsight because they are mistaken for signs of normal ageing or the stresses of a busy lifestyle. Owing to the diversity of diseases that cause dementia, the symptoms – while frequently overlapping – can vary substantially. Most commonly, they include some combination of memory loss, problem-solving difficulties, personality or behavioural changes, or trouble finding words. Specifically, however, depending on which brain area the underlying disease affects, the range of symptoms can be different.
Dozens of conditions are known to cause dementia, with the most common being Alzheimer’s disease. Other common types include frontotemporal lobar degeneration (FTD) and dementia with Lewy bodies. It’s worth noting that many different diseases lead to cognitive impairments, such as Huntington’s or prion diseases, including Creutzfeldt-Jakob disease (also known as Mad Cow disease). Identifying the underlying condition and its prognosis helps people receive appropriate treatment and support.
For about a third of people living with dementia, there is more than one underlying cause, a state referred to as mixed dementia. This is particularly true for people over 80. Mixed dementia can affect the severity and progression of symptoms, requiring specific management strategies.
Vascular dementia
You may have heard about vascular dementia, often said to be the second most common cause of dementia. This is almost certainly incorrect and relates to a tendency to label people with dementia who have some vascular lesions on their brain scan as 'vascular dementia'. MRI scans showing vascular lesions due to hardening of the arteries are common in people over 60. Thus, the cause of dementia can be falsely attributed to the lesions' presence, when the real cause is Alzheimer's disease. This is not to say that vascular lesions in someone with Alzheimer's are entirely irrelevant. People who have Alzheimer's with large amounts of vascular pathology tend to have a somewhat faster progressing dementia than those without these lesions.
Alzheimer's disease
Named after the German psychiatrist Alois Alzheimer (1864-1915), Alzheimer’s disease is the most common form of dementia. It affects women slightly more than men, though the reasons for this are unknown. While Alzheimer’s typically affects older individuals, it can also occur in younger people, even as young as 30.
Alzheimer’s is characterised by a progressive loss of brain tissue and the accumulation of two abnormal proteins, amyloid-beta and tau. This protein build-up results in the formation of ‘plaques’ and neurofibrillary tau tangles, which are the defining hallmarks of the disease.
Its symptoms primarily affect memory, thinking and reasoning, personality, and behavioural changes. At advanced stages, people living with Alzheimer’s struggle to perform simple everyday tasks and become reliant on care and support. The onset of memory symptoms is the most common variant of Alzheimer’s disease and most often why people seek medical attention.
Varying symptoms
People with Alzheimer’s do not always see a doctor about their memory. They may seek medical advice about their vision or challenges with language. These symptoms are caused by different variants of Alzheimer’s disease.
For example, one variant is Posterior Cortical Atrophy (PCA), also called Benson’s syndrome. In PCA, degeneration affects the brain areas responsible for spatial perception, complex visual processing, spelling and calculation. It frequently affects younger people (typically in their 50s). Often, people with PCA go to the optometrist first, thinking they have a problem with their eyes or need new glasses. The combination of relative youth, strange visual symptoms, and often relatively preserved memory, make this variant of Alzheimer’s disease notoriously difficult to diagnose in its early stages.
Primary progressive aphasia (PPA) is another variant that slowly damages the brain areas that control language. People with PPA have language symptoms, most notably, difficulty retrieving words in conversation and mispronunciation.


Alzheimer's causes
Most cases of Alzheimer’s have no known cause and are considered sporadic, resulting from a combination of genetic, lifestyle and environmental factors. At least 70 genetic risk factors have been linked to its development, though the nature of their impact remains unclear. Apolipoprotein E (ApoE) is a key gene associated with Alzheimer’s disease risk.
The ApoE gene has three possible genetic variants (known as alleles): E2, E3 and E4. Each of us has two copies of this ApoE gene, one inherited from each biological parent. So, if a person inherits the E2 allele from one parent and the E3 allele from the other, they are said to be ApoE2/ E3. Inheriting a copy of the ApoE 4 allele from one parent increases the risk of developing Alzheimer’s disease while inheriting the ApoE4 allele from both parents (ApoE4/E4 homozygote) means the risk is even higher. Testing for ApoE status is typically not done clinically because having no copies of the ApoE4 allele does not mean one cannot develop Alzheimer’s. Moreover, inheriting an ApoE4 allele does not guarantee that a person will develop Alzheimer’s. Therefore, ApoE testing cannot definitively rule the disease in or out.
Not all genetic factors, like ApoE, work by modifying risk. There are also gene mutations that, if inherited, will cause Alzheimer’s disease. These are rare and typically affect people at much younger ages than non-genetic Alzheimer’s (even in their 30s). In these forms, inheriting the faulty form of the gene from one parent (known as autosomal dominant inheritance) will cause the disease. Recalling that people have two copies of most genes, one of which is passed to their offspring, the child of a parent carrying one of these genetic mutations has a 50% chance of inheritance. Thus, on average, half the members of kindred carrying one of these mutations will develop Alzheimer’s disease.
Alzheimer’s is a complex disease, and scientists are still working to identify all the genetic variants that contribute to Alzheimer’s risk. A small percentage of Alzheimer’s cases are caused by inherited mutations.
Three known genes, when harbouring mutations, cause Alzheimer’s disease: APP (amyloid precursor protein), PSEN1 (presenilin 1) or PSEN2 (presenilin 2). Mutations in any of these genes result in excessive accumulation of amyloid-beta, the formation of plaques, and the loss of neurons in the brain typical of Alzheimer’s disease.
Key genes in late-onset Alzheimer's
Everyone receives two copies of the ApoE gene, one from their mother and one from their father. ApoE has three common forms or variants and you can inherit any two:
ApoE 2: rare and may protect against Alzheimer's
ApoE 3: common and seems to play a neutral role
ApoE 4: the risk gene. One copy increases risk 4-fold. Two copies increase the risk 7-8 fold.


The biology of Alzheimer's
Amyloid-beta
The main component of the hallmark plaques seen as lesions in the brains of Alzheimer’s patients (above) is formed by a protein called amyloid-beta. Accumulation of this protein is a key component of many current hypotheses for the cause of Alzheimer's. Amyloid-beta is produced by neurons in the brain. Its exact function(s) are not well understood, but it does play a role in memory, neurogenesis (the creation of new neurons), and signalling between neurons. However, when the production of amyloid-beta, outpaces its clearance, it accumulates around and between neurons, forming clumps or plaques. As these plaques grow, they induce a loss of synaptic receptors, leading to impaired signalling between neurons and, eventually, their death. Amyloid-beta accumulation is also thought to facilitate and trigger the other key player in Alzheimer’s pathology: tau.
A potential challenge to the hypothesis that amyloid-beta is central to the pathogenesis of Alzheimer’s are recent trials of drugs aimed at removing the amyloid-beta plaques from the brain. The effect of removing these plaques still needs to be tested.
The role of tau
Tau is a protein that plays a vital role in maintaining the structure of neurons, especially their axons (the long cables that transmit signals). Various types of tau accumulation occur in a multitude of human degenerative brain diseases, from genetically inherited disorders to environmentally acquired problems, such as traumatic brain injury.
In Alzheimer’s disease, tau accumulates and forms clumps called neurofibrillary tangles. These tau clumps interfere with critical cellular functions, such as mitochondrial function (energy generation), protein degradation, and synaptic plasticity, ultimately resulting in the neuron’s death. Scientists are still exploring the interplay between tau and amyloid- beta as they potentially act together in the development of Alzheimer’s disease or influence each other’s pathology.

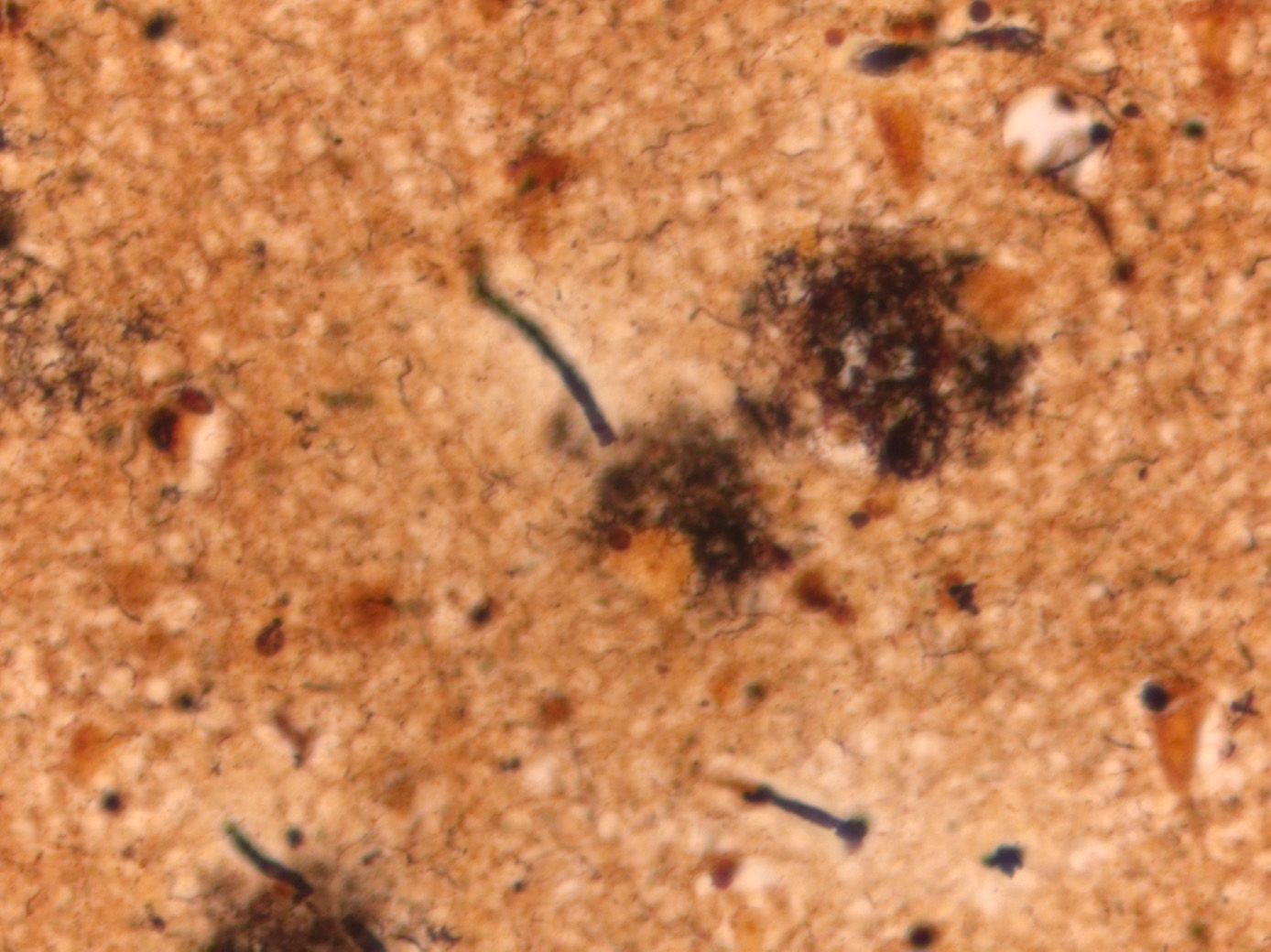
Amyloid plaques
Amyloid plaques
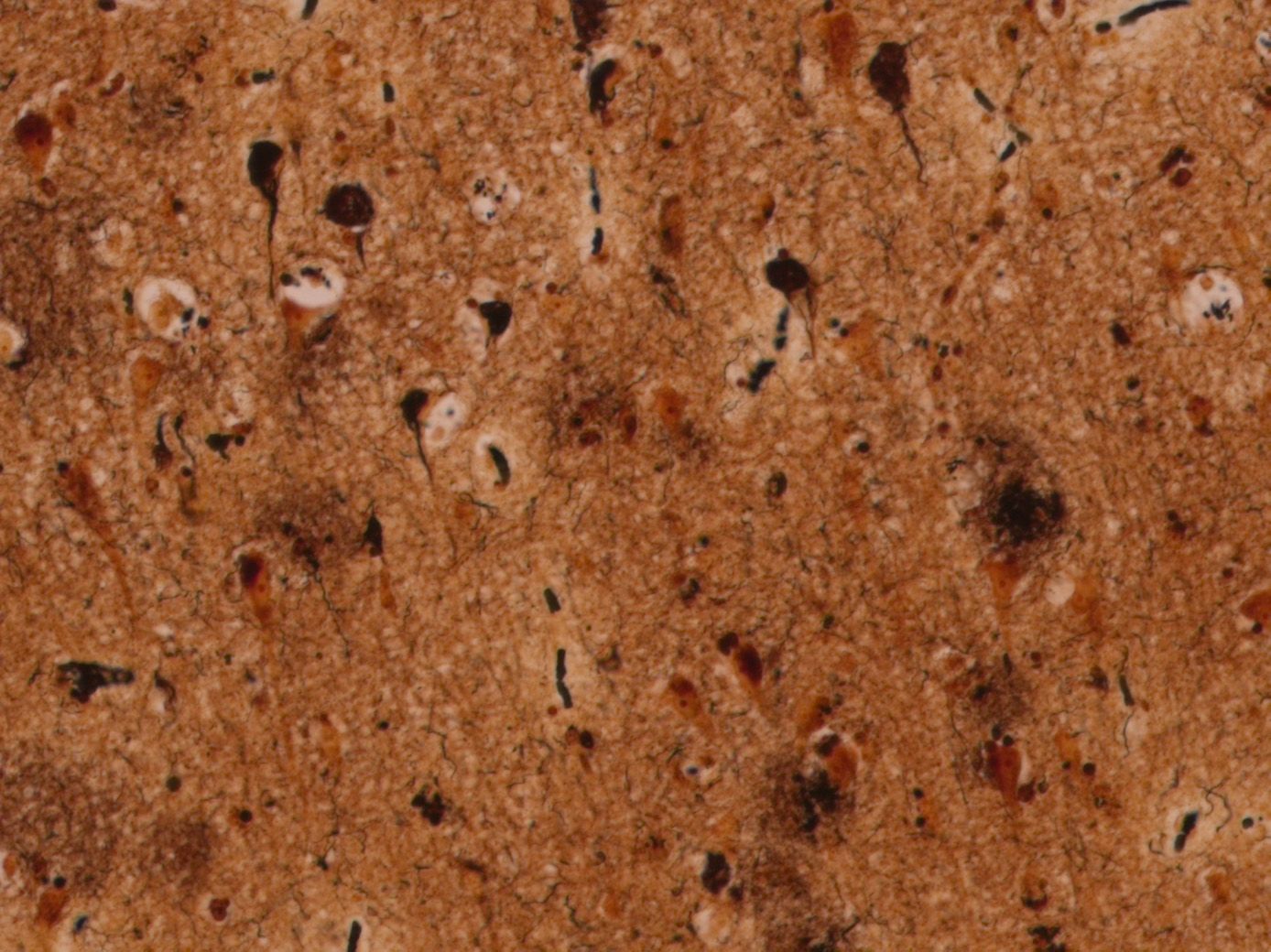
Tau tangles
Tau tangles
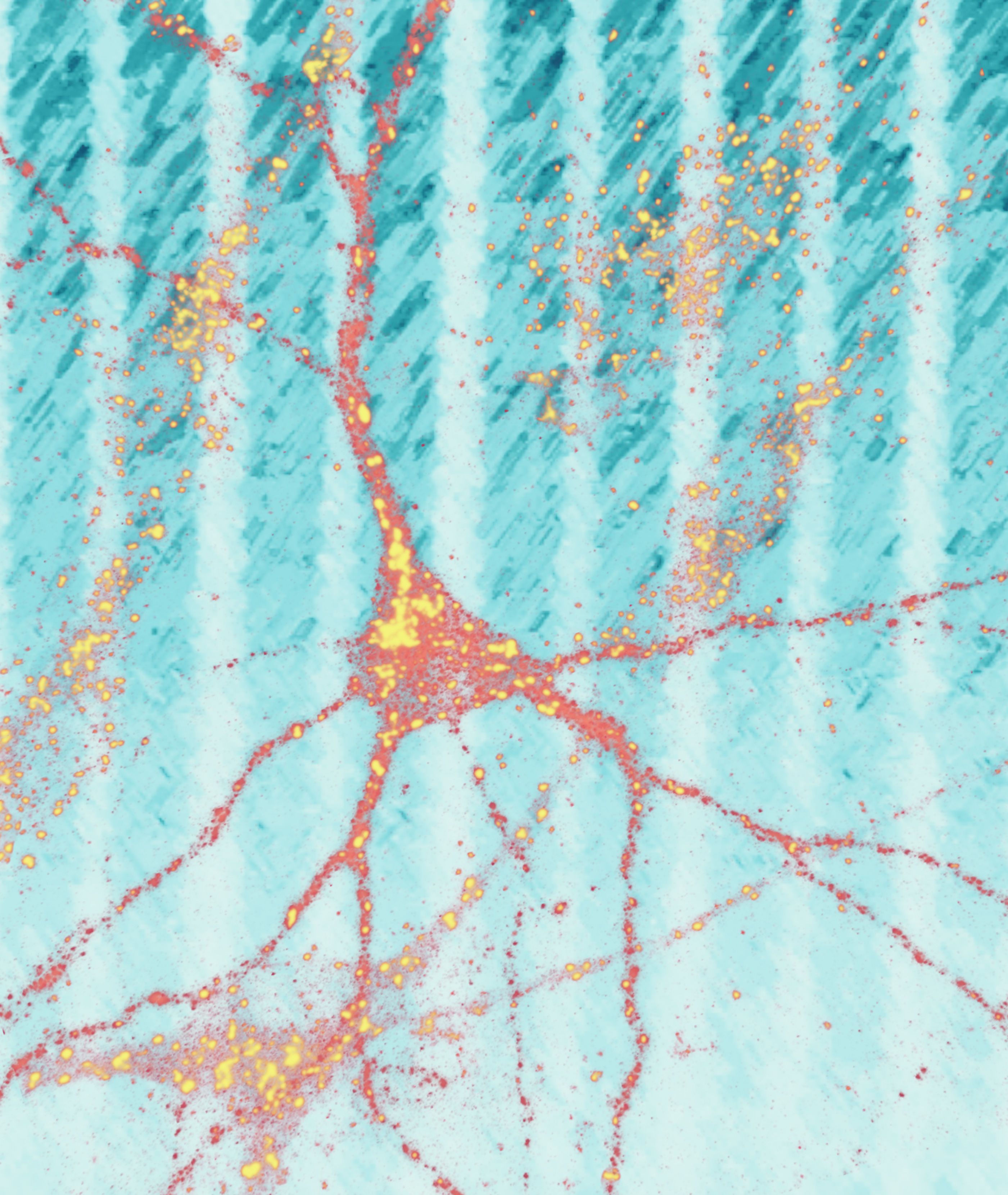
Stages of cellular changes

Amyloid-beta (yellow) starts to accumulate near neurons. Tau (black) starts to accumulate inside neurons.

Amyloid-beta plaques increase. Tau begins to deposit inside the neurons, causing damage.

Death of neurons occurs, severing connections with other neurons.
Frontotemporal lobar degeneration
Frontotemporal lobar degeneration (FTLD) is the second most common cause of dementia for people under 65 and the most common form for people under 60.
As with Alzheimer’s and other types of dementia, the brains of people with FTLD show loss of neurons with abnormal accumulations of certain proteins. The accumulating proteins differ between FTLD variants. Among these are TDP-43 (also found in motor neurone disease), tau (but different from the kind of tau found in Alzheimer’s), FUS (Fused in Sarcoma) and, most recently, TAF15.
Due to the affected brain areas’ role in cognitive functions like personality, emotion, behaviour, and language, people with FTLD present with differing symptoms. Unlike Alzheimer’s disease, memory is not usually affected. Clinically, FTLD presents either with behavioural changes, or with a language disorder.
People with the behavioural variant of frontotemporal dementia (bvFTD) initially present with personality and behaviour changes, e.g. in self-control or empathy. People with bvFTD typically do not recognise the changes in their own behaviour. These features are exceptionally challenging for their partners, with studies typically showing this form of dementia is associated with the highest levels of caregiver burden and stress. Their clinical symptoms can also overlap with other frontotemporal disorders, including progressive supranuclear palsy (PSP), corticobasal syndrome (CBS) and motor neurone disease (MND).
Language forms of FTLD, collectively known as primary progressive aphasia (PPA), typically manifest in one of two very different forms:
With semantic PPA (svPPA), people present with progressive loss of knowledge, including word meanings and object usage, due to degeneration of part of the temporal lobe. This manifests in their inability to name people and objects. This results in very fluent speech output that is lacking in specific content words. The deficit, however, is more than just a language problem of attaching names to objects or people. Knowledge of the underlying concepts is the core of the problem, meaning that people also lose understanding of what objects are rather than just their names.
With nonfluent PAA (nfvPPA), people have trouble speaking in fluent sentences due to degeneration of part of the left frontal lobe. In many ways, this is a mirror opposite to the language disorder of svPPA. Their comprehension of word and object meanings is mainly intact, but the syntax used to form sentences is faulty, manifesting as obviously nonfluent speech output. Pronunciation of words is also affected, as is the normal musicality of speech (prosody), so that conversation sounds strange and distorted. Although this condition can make people mute, it often spares other mental abilities. Although verbal communication is impossible, people can still care for themselves and perform tasks that do not involve speaking.
Though most people with FTLD have a sporadic disease, it is more likely to have a genetic basis than Alzheimer’s, with 10-15% of cases linked to identifiable genetic mutations. The most common is the C9orf72 gene, which also causes amyotrophic lateral sclerosis (ALS, also known as motor neurone disease or MND). In families harbouring this mutation, we find people affected by FTLD, motor neurone disease or both.
Other, less common genetic forms are associated with mutations in progranulin (GRN), microtubule-associated protein tau (MAPT), and a range of rarer genes.


Imaging of a patient with semantic variant primary progressive aphasia (svPPA)
Imaging of a patient with semantic variant primary progressive aphasia (svPPA)

Imaging of a patient with non-fluent variant primary progressive aphasia (nfvPPA)
Imaging of a patient with non-fluent variant primary progressive aphasia (nfvPPA)


Cortical thinning is not a feature of Dementia with Lewy bodies, so the MRI statistical map does not show any abnormalities
Cortical thinning is not a feature of Dementia with Lewy bodies, so the MRI statistical map does not show any abnormalities
Lewy body dementia
Lewy body dementia (LBD)—the second most common dementia type—is an umbrella term for Parkinson’s disease, including Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB). They share an underlying pathology characterised by the accumulation of the protein alpha-synuclein in neurons. These clumps of alpha-synuclein are visible under a microscope and are called Lewy bodies. As with all dementias, there is neuronal loss but, curiously, no prominent tissue loss, so the brain does not shrink (atrophy) as it does in other dementias. LBD incidence increases with age, with 75 the average age of onset.
LBD affects the brain with a range of different clinical manifestations and is associated with degeneration,in particular, of specific neurotransmitters. This dementia is characterised by problems with visual perception —including prominent problems with visual hallucinations —and trouble paying attention. There is a loss of function in the parts of the brain that process visual information. Loss of the neurotransmitter acetylcholine seems to be important in the development of these clinical features. Boosting acetylcholine is the mechanism of action of drugs such as donepezil and rivastigmine, which were developed for treating Alzheimer’s disease. Unsurprisingly, therefore, people with LBD often derive significant benefit from this class of medications.
Levels of the neurotransmitter dopamine also decline in LBD, and this gives rise to the movement disorder (slowness of movement, stiffness, tremors, balance problems) we know as Parkinson’s disease. People with this disease also often report symptoms of REM sleep behaviour disorder, a condition in which a person physically acts out dreams during sleep. This does not usually occur as our brain normally inhibits motor functions during REM sleep, but it is common among people with LBD. The disease also affects gut motility as a prominent early feature—in hindsight, when people are diagnosed with LBD, it is often noted that they had developed problems with constipation years beforehand. Pathology also affects the olfactory system, causing loss of normal smell and taste sensation.
What is Parkinson's disease?
Parkinson's disease is a progressive neurological condition that affects motor control and movement. Neuronal loss leads to symptoms that include tremors, stiffness, slowing of movement and trouble with balance. Parkinson's disease is intricately linked to dementia. Most people with Parkinson's go on to develop dementia symptoms, though it may not manifest until 10 to 20 years after the movement problems and initial diagnosis. Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB) are almost certainly the same disease. The dementia syndrome in each is indistinguishable from the other, as is the pathology in the brain. The only difference is that if the disease starts with a movement disorder, it called PDD, while if the dementia comes first, it is DLB.
LATE
Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE) is a recently classified dementia type in people predominantly over 80. LATE symptoms are like Alzheimer’s in that prominent memory problems are the hallmark. On its own, LATE progresses more slowly than other dementias, but about half of those with signs of LATE also had Alzheimer’s protein build-up, suggesting having more than one contributes to a faster decline.
Currently, LATE does not yet have a symptomatic diagnosis. Researchers identified LATE based on autopsy samples that revealed abnormal TDP-43 clusters, which are also involved in motor neurone disease (MND) and frontotemporal lobar degeneration (FTLD). However, LATE is not associated with MND or FTLD. This highlights a particularly curious phenomenon in degenerative brain diseases: specific protein aggregations can occur in unrelated diseases. This is particularly true of the protein tau, a feature of numerous degenerative diseases. Researchers are investigating ways to diagnose LATE in living people.

Staining techniques
Staining techniques are scientific methods that apply stains to tissue to study its cells and structure. Neuropathologists use staining to identify changes in the brain and patterns related to dementia types. As technology advances and researchers invent new stains, they compare what they find using stains on autopsy tissue to reach a consensus.
LATE was recently characterised using this approach. Advances in staining are also the reason for Alois Alzheimer’s name being associated with the most common form of dementia. That people can develop dementia was hardly a discovery at the beginning of the 20th century when he was working. However, the development of a new stain meant that Alzheimer could visualise the protein aggregates that characterise the disease for the first time.




Bridging the gap between brain and behaviour
Professor Gail Robinson
One obstacle to developing and delivering targeted dementia treatments is its complex nature. Dementia’s varying symptoms make early and accurate diagnosis of its different types and distinguishing these from other neurological disorders difficult.
Professor Gail Robinson, a clinical neuropsychologist and researcher who leads the Cognitive and Clinical Neuropsychology group at QBI and the UQ Neuropsychology Research Clinic, is addressing this challenge by combining theoretical neuroscience and clinical practice to bridge the gap between brain structure and function and behaviour.
Focusing primarily on the brain-behaviour relationships involved in frontal lobe functions (e.g., executive control functions, language generation), Professor Robinson and her team aim to develop new cognitive assessment and clinical diagnostic tools to identify more precise indicators for earlier dementia detection and improved diagnostic accuracy, which would allow for more effective management strategies.
There is no single test for diagnosing dementia. A diagnosis is usually reached using a combination of cognitive assessments that can measure a person’s challenges with thinking, learning, memory, and decision-making, a medical history review, physical and neurological tests and brain imaging.
The biggest challenge in dementia research is that there are many different types. It can manifest in symptoms like memory changes and in other areas of cognition, like language, executive functions and vision. This is where ‘precision phenotyping’ is needed at the behavioural and molecular level.
“My research is focused on understanding brain-behaviour relationships, such as what part of the brain is critical for creative thought.”
"For instance, understanding how we, as healthy individuals, formulate an idea and how we translate that idea into spoken language. In people with dementia, a disturbance in this process can manifest in problems with conversational speech. We focus on improving the detection and rehabilitation of these thinking problems.
“For example, we have shown that cognitive interventions and/or brain stimulation over the left frontal cortex improve spoken language production in healthy adults and those with neurological disorders, including dementia.
“I am also involved in several multidisciplinary research projects focused on ageing and dementia, for example, leveraging advanced imaging technology or developing new tools to optimise the early and accurate diagnosis of frontotemporal dementia.“
“A wonderful and exciting new project in collaboration with RMIT and supported by Dementia Australia is the “TuneChair”, a lounge chair modified to send low-frequency vibrations through the chair while people sit in it listening to peaceful music. My research investigates its benefits for cognition and mood in both healthy older adults and those with dementia.”
NEUROPSYCHOLOGIST: A neuropsychologist studies how the nervous system's (including the brain) structure and function relate to behaviour and cognitive skills. They assess cognitive abilities, such as memory or problem-solving. Neuropsychologists work with people who have Alzheimer's disease, evaluating how these conditions affect a person's ability to think, learn, remember and perform daily tasks.